

Background: Myelodysplastic syndromes (MDS) are a group of clonal stem cell neoplasms, presenting with cytopenias. Diagnosis relies on peripheral blood counts and bone marrow morphology, classifying MDS patients by percentage of blasts and number of cell lineages affected. Although half of the patients show normal karyotypes, cytogenetic profiles allow risk stratification (IPSS-R). A number of recurrently mutated genes were identified (Haferlach et al, Leukemia 2014; Papaemmanuil et al, Blood 2013), but in the current WHO classification gene mutations are still only mentioned as a footnote. However, with increasing sequencing capacities the spectrum of investigated genes is also becoming broader. 85% of MDS patients carry at least one mutation at diagnosis but the median number of mutations per patient is low, illustrating that - genetically speaking - MDS is a quite simple disease.
Aim: To determine the actual gain of genetic information with increasing NGS panel sizes, starting with small panels of diagnostic guidelines to the complete coding region (=exome) covered by whole genome sequencing (WGS).
Patients and Methods: The cohort comprised 588 patients (234 female and 354 male) with a median age of 72, diagnosed as MDS by cytomorphology and cytogenetics following WHO classification. All patients were previously investigated by next generation deep sequencing with a mean read depth of 1,066x (Haferlach et al, Leukemia 2014) and WGS. WGS libraries were generated with the TruSeq DNA PCR-Free Kit and sequenced with 150 bp paired-end reads on NovaSeq or HiSeqX instruments with a mean coverage of 106x (Illumina, San Diego, CA). Variant interpretation was performed by considering ClinVar, Cosmic, functional impact predictors and a population frequency in gnomAD ≤0.00005. No or discordant information led to classification of variant of uncertain significance (VUS), considering only protein altering mutations.
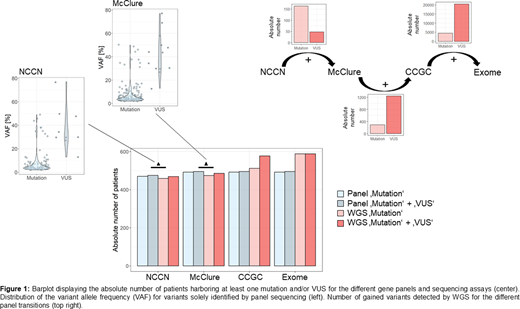
Results: The targeted sequencing and WGS data was analyzed for 2 and 4 virtual gene panels respectively: the 12 genes as given in the MDS NCCN guidelines with a incidence >5% (Version 2.2020; "genes frequently somatically mutated in MDS"), the 34 gene panel summarized in McClure et al. (J Mol Diag 2018; "minimum recommended list for management of the most chronic myeloid neoplasms"), the 723 cosmic cancer gene census (CCGC, v91) panel and finally the exome (figure 1). All of the genes from the NCCN panel were mutated in at least one patient, summing up to an overall count of 1,058 mutations. 81% of patients carried at least one mutation, with a median of 2 mutations per patient. Only a few VUS occurred (n=50) based on our classification strategy. Extending the analysis to the McClure panel revealed 196 additional mutations in 18 additional genes, increasing the number of patients with a least one mutation by 18 to 492 (84%). The number of VUS rose to 73. Reviewing the WGS data for the two panels showed a congruence of 86.8% for the identified mutations. Mainly small clones (VAF <15%, n=151) were missed due to the lower sensitivity of WGS. Addressing the cosmic cancer gene set only 290 mutations were detected on top. The number of patients with at least one mutation increased marginally to 512 (87%), while a large number of additional VUS was detected (n=1,240), illustrating the difficulty in classifying this data. The phenomenon was even more pronounced in the exome data set with a remarkable increase of detected VUS by 20,540. All patients harbored at least one mutation and the number of recurrently mutated genes was elevated to 770. However, most of the gene mutations were unique. The median number of mutations per patient was 10, found in 3,603 different genes, including LUC7L2 and CUX1, located on chromosome 7q, CSNK1A1, located on 5q, and various transcription factors and RAS pathway related genes.
Conclusions: MDS cases can be investigated by a small diagnostic gene panel. The higher sensitivity of targeted deep sequencing allows detection of small clones, relevant for cases with CHIP or CCUS. A large gene panel shows only small gain of clinically relevant information but many additional VUS. The restriction to a panel containing less than 30 genes leads to cheaper sequencing costs and especially less time for bioinformatics and clinical interpretation of findings. Therefore, the choice of a small but well selected gene panel is more favorable than large sequencing efforts.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal